| |
It is highly desirable to theoretically predict the molecular fluorescence efficiency in order to design highly efficient organic light-emitting materials, molecular switches as well as bio-sensing molecules. The light-emitting efficiency is determined by the competition between the radiative and non-radiative decay processes. While the radiative process can be understood through the Einstein’s relationship between spontaneous and stimulated radiation processes, the non-radiative decay remains a great challenge for theoretical chemistry, which consists of an essential difficulty for predicting the light-emitting efficiency. The conventional theory was developed based on the assumption that the ground state and the excited state possess the same harmonic parabola except a rigid shift in origins. Such approximation can result in about 2 to 3 orders of magnitude deviation in estimating the rate constants for organic soft molecules. Thus it does not allow a quantitative prediction of light-emitting efficiency. Furthermore, it can not reasonably explain the temperature dependence of the fluorescence efficiency dependence on the temperature.
Prof. Zhigang Shuai of the CAS Key Laboratory of Organic Solids of the Institute of Chemistry and coworkers have recently developed a new formalism for determining the nonradiative decay rate constant by taking into accounts of multimode mixing Duschinsky rotation effects, which can describe the difference in potential energy surfaces between the excited state and the ground state. The new formalism is fully analytical, and is implemented in the first-principles framework of time-dependent density functional theory (J. Chem. Phys. 2007, 126, 114302). Furthermore, they have implemented a computational scheme for the radiative decay based on Einstein’s spontaneous radiation process coupled with molecular vibrational levels distributoion. The theory has successfully explained the completely different photophysical behaviors for the two isomers of tetraphenylbutadienes, and revealed the origin of the exotic aggregation induced emission phenomena for one isomer. Especially the temperature dependence of the fluorescence can only be explained under the current new formalism with Duschinsky rotation effect. It is a parameter-free theory, namely, once a molecular structure is given, the molecular fluorescence efficiency can be calculated. As an independent verification of the theory, Z. Shuai and coworkers also applied this formalism for the photophysically known molecules, the three conformers of diphenylbutadienes. It is found that both the calculated radiative and non-radiative decay rates are in good agreements with the measurements for the trans-trans isomer. Since the cis-trans and cis-cis isomers are non-emissive, the rates constants are impossible to be measured. However, these can be calculated, and eventually the obtained quantum yields for various processes agree well with the experiments. This work has been published in J. Am. Chem. Soc.(2007, 129, 9333-9339).
This new formalism will play an important role in molecular design of organic light-emitting materials. Right after the publication of the work, it was highlighted under the title “Luminescent or nonluminescent? Predict the fate of an excited chromophore from first principles” at the homepage of the American Chemical Society (www.acs.org, Aug. 27, 2007) in the “Heart Cut Paper” weekly column. It is pointed out that “The excited state of a molecule can decay to the ground state via radiative and nonradiative transitions. Whereas the radiative decay process can be followed spectroscopically, the nonradiative decay process is difficult to measure and hence not well understood. Z. Shuai and coauthors at the Chinese Academy of Sciences (Beijing) and Beijing Normal University tackled nonradiative decay with a first-principles approach and established a formalism that can predict the outcome of fluorescence processes of ‘large’ molecules…”
This work was supported by the China Ministry of Science and Technology through the 973 program, by the National Science Foundation of China, as well as by the Chinese Academy of Sciences.
J. Am. Chem. Soc.(2007, 129, 9333-9339).
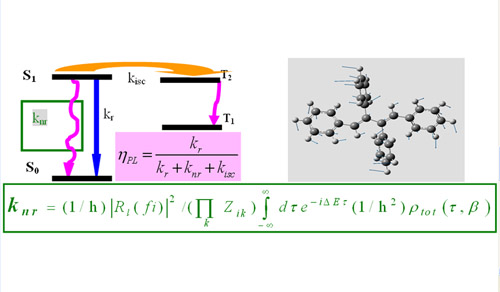
The molecular excited state kinetics and the fluorescence efficiency can be described by the simplified Jablonki diagram. However, it remains a great challenge for theoretical chemistry to predict the rate constants from first-principles. This research group has derived for the first time the fully analytical formula (see above). The upper right panel shows the microscopic processes of how molecule dissipates its electronic energy through nuclear vibrations.
, |
|